The MEAP-VHSV database contains records that provide background information and genetic sequencing data for individual isolates of the fish virus Viral hemorrhagic septicemia virus (VHSV). The database focuses on VHSV isolates collected from free-ranging fish throughout the Great Lakes region in North America from 2003 to the present. To the best of our knowledge, it includes all available virus isolates collected from Great Lakes fish since the emergence of VHSV in 2003. In the future we will add VHSV isolates from the west and east coasts of North America, and a small number of representative isolates from Europe and Japan. The purpose of this database is to provide access to detailed data for all parties interested in North American VHSV molecular epidemiology, such as fish health professionals, fish culture facility managers, and academic researchers. The database has flexible search capabilities and generates various output formats, including tables and maps, that we hope will assist users in developing and testing hypotheses about how VHSV moves across aquatic landscapes and changes over time. By engaging the expertise of the broader community of colleagues interested in VHSV, our goal is to enhance the overall understanding of the emergence of VHSV in the North American Great Lakes.
VHS virus is a well known fish pathogen with a very broad host range including species in many diverse fish families. Taxonomically VHSV is in the genus Novirhabdovirus, within the virus family Rhabdoviridae, which includes other well known animal viruses such as rabies virus and vesicular stomatitis virus. VHSV was originally identified as a major pathogen of trout farms in Europe, but it is now clear that there is a large multi-species marine reservoir of VHSV in both the North Pacific and North Atlantic Oceans. VHSV can be transmitted either horizontally through waterborne virus, or vertically as egg-associated virus. Infection can result in lethal internal and external hemorrhage, and epidemics may reach up to 90% mortality depending on various host, virus, and environmental factors. VHSV is a significant impediment to rainbow trout farm culture in Europe, and epidemics among wild sardines and herring have been documented off the Pacific coast of North America. Prior to 2003 VHSV had never been detected in fish from the Great Lakes region, but since then the virus has emerged dramatically, causing several large, multi-species epidemics among free-ranging fish at numerous locations in 2005, 2006, and 2007. Detections of VHSV in Great Lakes fish continue to the present, and it has been isolated from 28 different fish host species. Geographically VHSV has been detected in all five Great Lakes, and in several inland lakes within the region, but to date it has not been detected in Great Lakes aquaculture facilities.
Before the emergence of VHSV in the Great Lakes, molecular epidemiology of VHSV field isolates defined four major phylogenetic genogroups designated genotypes I-IV. Genotypes I, II, and III occur in marine fish in waters surrounding Europe and the United Kingdom, and freshwater trout farm isolates fall into subgroup Ia within genotype I. At that time North American VHSV from the Pacific coast was designated genotype IV. Characterization of Great Lakes VHSV has shown that it is most closely related to VHSV from the west coast of North America, but with enough separation to merit designation of West Coast isolates as subgroup IVa, and Great Lakes isolates as subgroup IVb
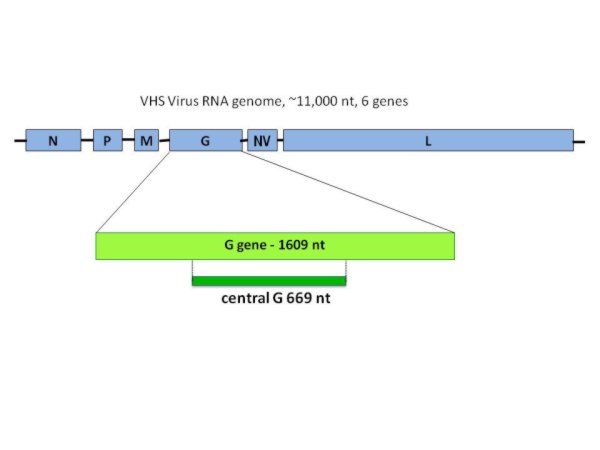
VHSV has a single-stranded, negative sense RNA genome of approximately 11,000 bases. This genome has six genes, which are ordered along the genome as 3' N (nucleocapsid), M (matrix), P (phosphoprotein), G (glycoprotein), NV (non-virion), and L (large polymerase)-5'. The G gene is approximately 1610 nucleotides (nt) long and encodes the only protein on the outer surface of the enveloped virus particles. Standard genetic typing was done by sequencing a variable 669 nt region in center of the G gene, referred to as the "central G" (Figure 1). The central G sequence has been determined for all isolates in the database. Each different central G sequence is referred to as a "genetic type", and any virus isolates with that exact same central G sequence would be grouped within that genetic type. Thus, some genetic types represent single isolates, and others may represent a large number of isolates. A universal nomenclature system has been developed to describe these genetic types. The "Universal Sequence Designator", or USD, for VHSV central G genetic types is written as "vcGXXX", where XXX is a randomly assigned 3 digit number identifying a specific central G sequence. For example, vcG001 is the most common central G sequence (genetic type) and it is found in the majority of the isolates, and vcG008 is found in only one isolate.
The database can be searched either by nucleotide sequence or by USD to identify all isolates with identical genetic types. The value of the USD system is that it allows us to talk about what the genetic typing data tells us about VHSV diversity and epidemiology. To date there is very little genetic diversity among Great Lakes VHSV isolate central G sequences. Isolates with the same USD are likely more closely related than isolates with different USDs, Isolates with the same USD may indicate epidemiologically linked isolates, and they should be considered for spatial, temporal, or host links. Although identical USDs indicate a relatively recent common ancestor, it is important to remember that the central G region is short and sequence differences may well occur outside this region. When isolates have different USDs the sequence differences function as tags or markers that can be useful for tracking the virus over aquatic landscapes or through time. Single nt differences suggest close relationships, and may represent relatively recent divergence by mutation compared with isolates that have multiple nt differences. As a simple example of interpretation, if two isolates from the same collection location differ in central G sequence by 1 nt, they could have arisen either by a relatively recent mutation that occurred at that location to create a variant, or by independent introductions of the two different types. However, if two isolates from the same location differ by 5 nt in the central G, that more strongly suggests two introduction events. It is important to keep in mind that without additional testing sequence differences at this level cannot be assumed to indicate any biological or phenotypic difference between isolates.
The methods used here for genetic typing of VHSV field isolates were developed by Bill Batts in the laboratory of Jim Winton at the U.S.G.S. Western Fisheries Research Center (WFRC) in Seattle, Washington. Virus isolates and their associated background information were obtained from various state, federal, and academic fish health colleagues. A virus "isolate" is defined as the virus produced in cultured fish cells inoculated with a fish tissue sample collected from a specific fish, or pool of fish, at a specific time and location. The sequences in this database were generated in the Winton and Kurath laboratories at WFRC.
The isolates included here do not necessarily represent consistent geographic or temporal coverage of VHSV in the Great Lakes. We have included all available Great Lakes VHSV isolates for genetic typing. Spatially some collection sites are represented by many isolates and some have only one or a small number. At present this database does not include VHSV isolates from North America outside the Great Lakes region, or from Europe or Asia. In the future VHSV isolates from the west and east coasts of North America will be added, along with a small number of representative isolates from Europe and Asia. Information on European VHSV can be found in the Fish Pathogens.eu database at http://www.fishpathogens.eu/vhsv. Coastal North American and Asian VHSV are described in published scientific literature.
Einer-Jensen K., Ahrens P., Forsberg R. & Lorenzen N. (2004) Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. Journal of General Virology 85, 1167-1179.
Elsayed, E., Faisal, M., Thomas, M., Whelan, G., Batts, W., & Winton, J. (2006). Isolation of viral haemorrhagic septiceaemia virus from muskellunge, Esox masquinongy (Mitchill), in Lake St Clair, Michigan, USA reveals a new sublineage of the North American genotype. Journal of Fish Diseases 29, 611-619.
Jonstrup, S.P., Gray, T., Kahns, S., Skall, H.F., Snow, M., and Olesen, N.J. 2009. FishPathogens.eu/vhsv: a user-friendly viral haemorrhagic septicaemia isolate and sequence database. J. Fish Diseases 32, 925-929.
Meyers T.R. & Winton J.R.(1995). Viral hemorrhagic septicmia virus in North America. Annual Review of Fish Diseases 5,3-24.
Smail, D.A. (1999). Viral Haemorrhagic Septicaemia. Pages 123-146 In Fish Diseases and Disorders, Volume 3, Viral, bacterial, and fungal infections. P.T.K. Woo & D. W. Bruno, eds. CAB International, New York.
Snow M., Bain N., Black J., Taupin V., Cunningham C.O., King J.A., Skall H.F., & Raynard R.S. (2004). Genetic population structure of marine viral haemorrhagic septicaemia virus (VHSV). Diseases of Aquatic Organisms 61, 11-21.
Thompson, T., Batts W.N., Bowser, P., Faisal, M., Philips, K., Garver, K., Winton, J., and Kurath, G. Low genetic diversity in the emergence of viral hemorrhagic septicemia virus in the North American Great Lakes Region. In preparation